Introduction
Flex-sweep is a convolutional neural network–based method able to detect a wide range of selective sweep signals, including those thousands of generations old, from single-species genomic data, while being robust to background selection. It was primarily designed to be flexible in terms of the types, strengths, and ages of sweeps, requiring only a single phased population — without outgroups or admixture data — making it particularly useful for non-model species.
Flex-sweep 2.0 is a more general-purpose update that streamlines the workflow, vastly speeds up summary-statistic computation, relaxes CNN constraints by supporting custom architectures, and enables ancestral-state polarization, while being highly customizable and robust to model mis-specification. Flex-sweep now scales to thousands of simulated training scenarios and enables genome-wide inference on a standard workstation rather than an HPC.
Similarly to the first version, Flex-sweep works in three main steps: simulation, summary statistics estimation (feature vectors), and training/classification. Once installed, you can access the Command Line Interface (CLI) to run any module as needed.
Pipeline overview
Simulations
The simulations module takes advantage of demes to simulate custom demography histories. The software includes a pre-compiled discoal binary to avoid external dependencies (a custom binary path can be provided if needed). Prior parameters are flexible across different distribution configurations and can be set via the Python API or CLI without manually editing custom input files.
Feature vector estimation
The feature vector module was fully refactored for speed and traceability. Summary statistics are estimated using Numba and NumPy vectorisation for all statistics except \(nS_L\) and \(iHS\), which rely on scikit-allel functions. All outputs rely on Polars DataFrames, removing the large number of intermediate files required by the previous version and substantially reducing RAM consumption.
The software directly reads VCF files — no conversion to hap/map format is needed. When using recombination maps, genetic positions and recombination rates are interpolated automatically.
The refactored version includes optimised implementations of iSAFE, DIND, hapDAF-o/s, Sratio, highfreq, lowfreq, as well as the custom HAF and H12 statistics from the original Flex-sweep paper. Additional statistics have been added (see Advanced usage for the full list).
Users can select any custom combination of the available statistics via the
stats= argument, choosing only those most informative for the organism and
sweep type under study. The software also supports user-defined genomic
intervals and window sizes, enabling feature extraction across highly
customisable regions. This design makes it straightforward to reproduce the
feature set of other tools (e.g. diploS/HIC) or to define a tailored feature
vector without writing custom code.
Normalization
When a high-quality recombination map is available, Flex-sweep normalises both SNP-based and windowed statistic within bins defined by the empirical recombination distribution. For windowed statistic each input is standardized within bins defined by the joint combination of window position, window center, and local recombination rate, extending the normalization scheme to explicitly account for recombination rate heterogeneity across the genome:
where \(\pi_i\) is any given statistic and \(\mu^{(w,c,r)}\), \(\sigma^{(w,c,r)}\) are the mean and standard deviation computed across all windows sharing the same window size \(w\), center \(c\), and recombination rate bin \(r\).
For SNP-based statistics, normalization is performed analogously but conditional on derived allele frequency (DAF) and local recombination rate. Specifically, each SNP-level statistic is first standardized within bins defined by the joint distribution of DAF and recombination rate:
where \(\pi_i\) is the given statistics, \(f\) denotes the derived allele frequency bin of SNP \(i\), and \(\mu^{(f,r)}\) and \(\sigma^{(f,r)}\) are the mean and standard deviation computed across all SNPs falling within the same DAF bin \(f\) and recombination rate bin \(r\). Finally, once each window and center combination is defined we normalized using the same approach described above.
Training and prediction
Because feature vectors are flexible to any combination of statistics, genomic center, and window size, the new API provides an automatic interface for custom CNN architectures — supporting both 1D and 2D CNNs. State-of-the-art haplotype rearrangement strategies are also available for working with raw haplotype matrices (see Advanced usage).
Domain-Adaptive Neural Network (DANN)
Flex-sweep is now more versatile for analysing non-model organisms where the quality or availability of simulated parameters — such as mutation rate, recombination rate, and demography — is limited. It extends the method to work with the Domain-Adaptive model proposed by Mo, Z. and Siepel, A. 2023. The Domain-Adaptive Neural Network trains not only on labelled simulated data but also incorporates unlabelled empirical data during training, learning a shared representation that is highly predictive for the classification task but uninformative about the domain (simulated vs. real). In the case of CNN approaches, models trained under unrealistic demography, recombination, or mutation landscapes can easily confound sweep prediction due to overfitting of artifacts; the DANN is explicitly designed to account for and mitigate this mismatch.
Note
When working with extremely out-of-range demographics (e.g. training over constant population sizes) or simulated parameters, domain-adaptive training may still perform worse than the plain CNN.
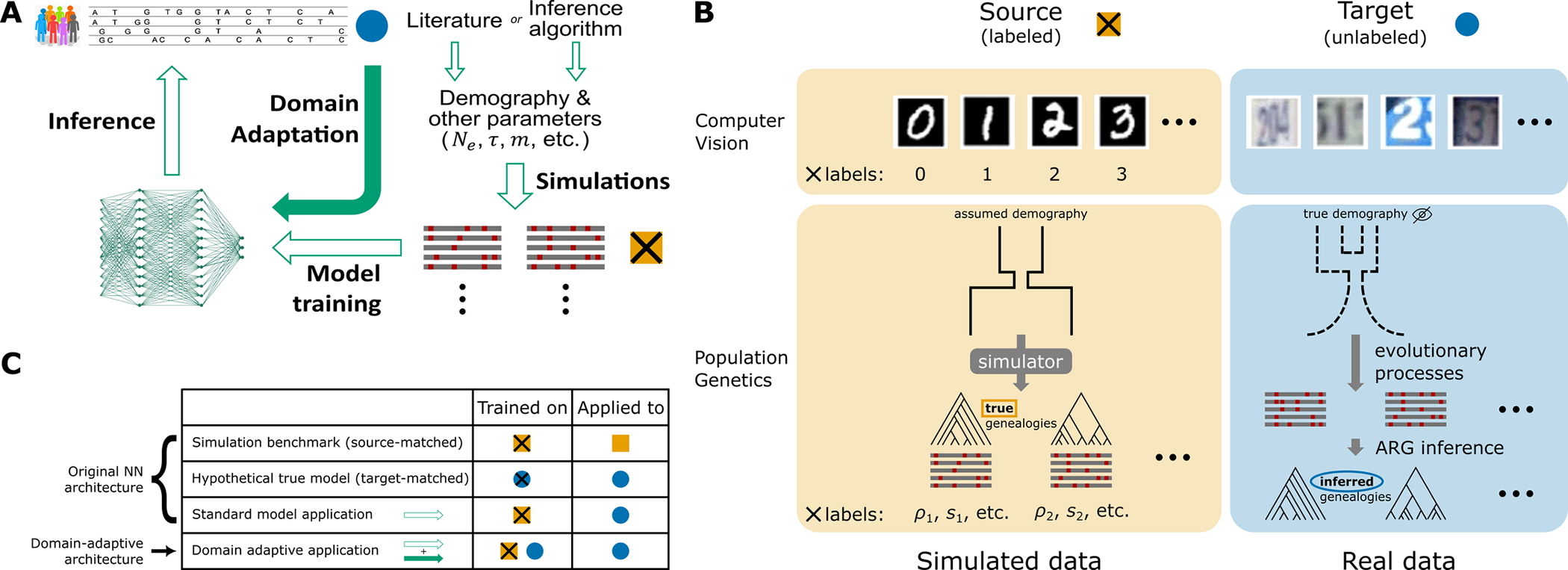
Figure extracted from Mo, Z. and Siepel, A. 2023.
Summary of neural networks, domain adaptation architecture, input data and benchmarking in the context of population genetic inference. Flex-sweep takes the same approach but Source and Target outputs are feature-vector images instead of genealogies.
Polarization
Flex-sweep refactors and extends est-sfs to support straightforward allele polarization. Given a focal-species polymorphism dataset (VCF) and a Multi-Alignment Format (MAF) file, the pipeline:
Subsets up to three outgroup species from the alignment.
Extracts aligned outgroup bases at polymorphic sites.
Computes per-site posterior ancestral-state probabilities following Keightley and Jackson 2018.
These posteriors are used to probabilistically assign ancestral and derived states, enabling downstream selection scans that are robust to ancestral mis-specification. .. This design is intended to take advantage of large .. comparative genomic resources such as the .. Zoonomia Project.
Genome-wide outlier scan
In addition to the CNN/DANN prediction pipeline, Flex-sweep includes a standalone genome-wide outlier scan that works directly from VCF files without any simulation or training. Each statistic is computed at its native genomic resolution and ranked against its empirical distribution to produce a p-value:
where a small \(p_i\) indicates a high-scoring outlier window. Statistics can be normalised jointly by allele frequency and recombination rate when a recombination map is provided. This provides a fast, assumption-light complement to the CNN approach, or a standalone analysis when simulations are not available. See Outlier scan for full documentation.
Rank and enrichment
A rank algorithm post-processes sweep probabilities and associates them with any genomic element in a BED file, using polars-bio to estimate genomic distances efficiently. See CNN prediction for usage.
To assess whether top-ranked genes or elements are sweep enriched, Flex-sweep implements the enrichment pipeline described in Enard et al. 2020 (see Enrichment analysis for details).
Recommended requirements
Flex-sweep runs now on a standard workstation. The minimum requirements depend mainly on the species genome size and sample size. The examples in this documentation use the YRI population (n = 108) from the 1000 Genomes Project and were run on the following configuration:
Pop!_OS 22.04
AMD Ryzen 9 PRO 5945 workstation (24 cores)
1 TB M.2 NVMe SSD
64 GB RAM
NVIDIA GeForce RTX 3080